




ChemBio3D Ultra是一款非常好用的化学绘图软件。专业的化学绘图工具ChemBio3D Ultra。您可以使用此软件来帮助用户设计各种化学图形,例如设计化学分子图,设计化学反应图,可以构建,可视化和分析化学结构的3D模型,还可以将ChemBio3D模型导入桌面发布工具或将它们显示在Web上,ChemBio3D随附GAMESS,并附带量子量,提供额外的计算引擎,可以分析软件中的生物学数据;提供CS MOPAC计算模块,CSMOPAC原子和分子的半经验计算以确定分子结构和特性的详细信息。提供了两个CSMOPAC选项:CSMOPACUltra和CSMOPACPro。 CSMOPACU包含MOPAC的所有功能,并且只能用作可选插件。 CSMOPACUltra支持高级功能,例如MOZYME和PM5方法。
安装方式:
1.打开ChemBio3DUltra140_Trial.exe软件并直接开始安装,单击“下一步”。
2.提示软件安装协议的内容。阅读协议后,单击“下一步”,您可以将协议拖到末尾,然后向下滚动到末尾。
3.软件的安装地址为C:\ Program Files(x86)\ CambridgeSoft \ ChemOffice2014
4.这是软件的安装内容,单击“下一步”。
5.相关插件的安装,默认是可以的
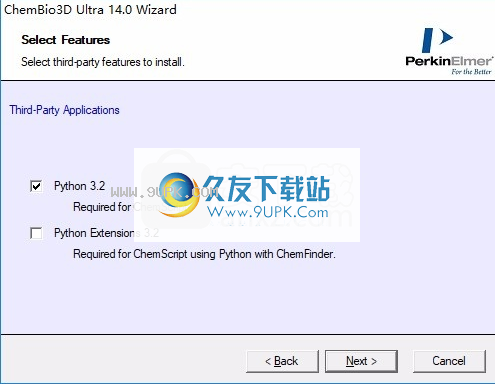
6.对于软件的安装内容,请直接单击安装以执行安装。
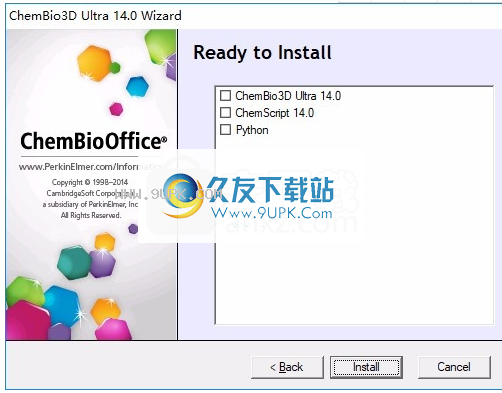
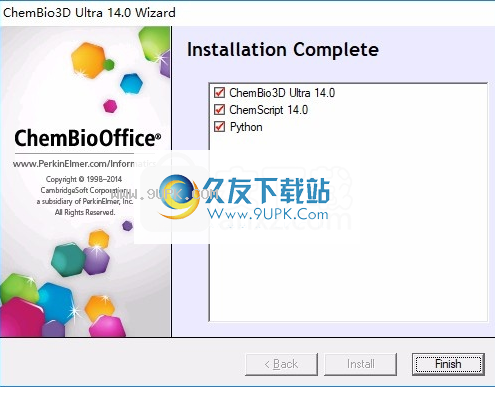
7.提示这三个项目的安装已结束,单击“完成”以结束。
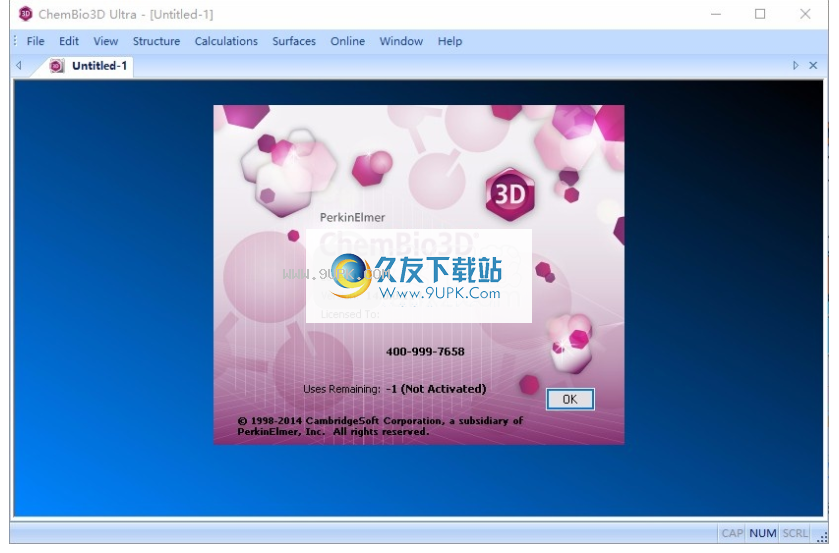
8.打开chembio3d软件,可以正常使用,该软件为试用版
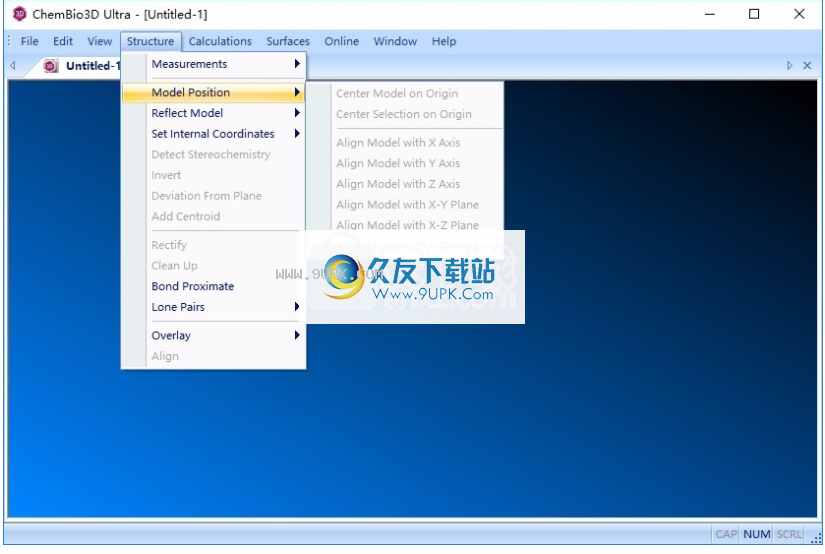
9.原点中心模型,原点中心选择,X轴Alian模型,Y轴Alian模型,将模型与轴对齐,并将模型与X-Y平面对齐
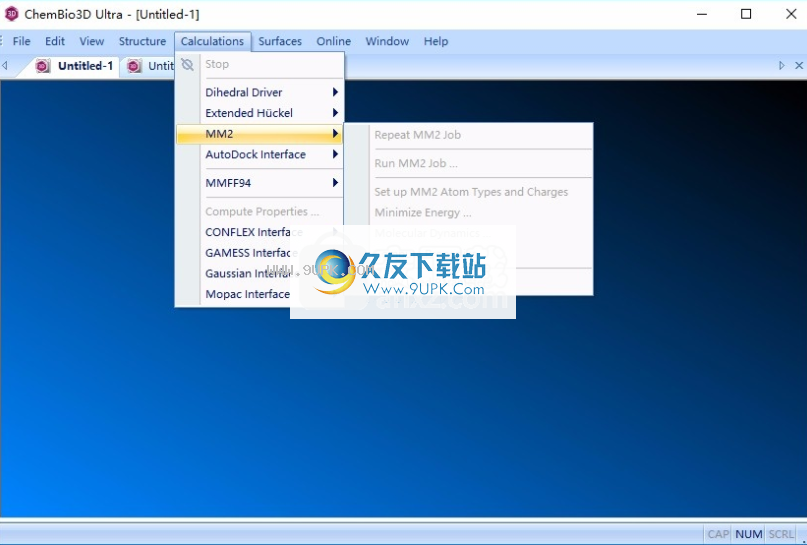
10.重复MM2作业,运行MM2作业,设置MM2 Atom类型和cos
t,降低能量,分子动力学,计算特性,显示使用的参数
软件特色:
1.视觉造型
通过3D建模,可视化和计算功能,科学家可以更好,更快地确定研究重点,从而提高生产率并快速实现研究目标。学生可以使用ChemBio3D Ultra的正式版来更深入地了解分子的3D空间结构及其对分子性能的影响。
2.2D和3D模式结构互换
ChemBio 3D集成了ChemBioDraw,可以简化2D和3D模式之间的结构交换。科学家可以研究小分子和生化试剂的三维形状和特性,并使合成化学家与计算化学家之间的交流更加有效和开放。
3.丰富的应用界面
ChemBio 3D经济实用,完全可以满足大多数科学家的需求,并且还可以提供与其他应用程序的接口,以满足用户对更高级功能的需求。该程序可以在普通的桌面系统上运行,而无需配备高性能和高端图形功能的系统。
4.兼容多种程序,准确的预测结果
ChemBio 3D包括多个可选版本,例如半经验建模程序MOPAC,半经验和从头算高斯分子轨道计算,并且可以连接到从头算程序GAMESS和可以处理灵活配体的自动对接程序AutoDock 。
5.显示费用
ChemBio3D也显示在ChemDraw图中。 ChemBio3D将以形式显示带电原子。如果原子有不同的位置
软件功能:
1.生产结构。这是ChemBio3D Ultra 14.0作为化学绘图软件的第一个重要功能
2.三维旋转。立体声选项是动态演示功能。选择围绕X,Y或Z轴旋转后,按F5或Ctrl + F5旋转结构。
3.阅读ChemDraw结构信息。 ChemBio3D的三维信息和ChemDraw的平面结构可以相互转换。
4.定量结构。 ChemBio3D的“计算”菜单下有许多命令可用于量化结构并执行结构优化计算。
5.与其他软件兼容。 ChemBio3D Ultra 14.0具有良好的兼容性。除了与一系列ChemDraw软件兼容外,它还与多种量子化学软件兼容,并且可以组合使用。
官方教程:
解
本节介绍带有各种屏幕元素及其用法的图形用户界面(GUI)。
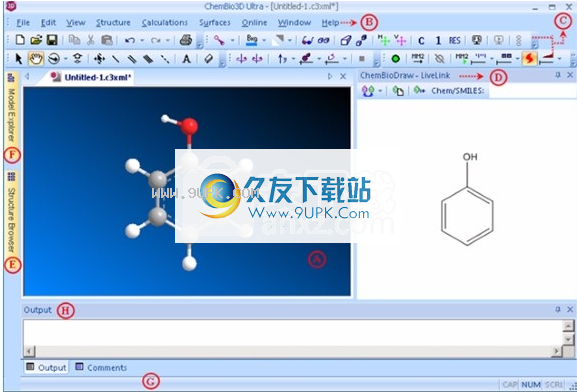
主屏幕:A)模型窗口; B)主菜单; C)工具栏; D)ChemDrawpaneltab; E)StructureBrowsertab; F)ModelExplorertab; G)状态栏; H)输出窗口。
模型窗口。在其中构建和查看模型的工作空间。主菜单。包括构建和查看模型,运行计算以及显示化学性质所需的所有工具。上下文菜单中还提供了许多菜单选项。工具栏。包含可以提供许多常用功能快捷方式的图标。您可以单击工具栏并将其拖动到屏幕上的任意位置。
ChemDrawpanel。
允许您在二维中创建或修改模型的图形。 StructureBrowser。用于浏览模型列表中的结构列表并查看每个窗口。模型资源管理器。显示模型的层次树表示。该模型可用于处理复杂的分子,例如蛋白质。状态栏。显示有关隐藏在模型区域中的原子的活动框架的信息。输出窗口。显示文本结果,例如计算结果。
模型窗口
您可以在模型窗口中构建模型。可以打开每个模型文件,也可以创建一个新选项卡。选择目录以激活您要查看的模型。文件名旁边的星号表示该文件自上次更改以来尚未删除。

模型浏览器
默认情况下,模型的爆炸风险将崩溃并显示为垂直对齐。将鼠标悬停在模型浏览器选项卡上时,将显示模型浏览器窗口,并显示当前显示模型的更高级树形表示形式。
样本文件
ChemBio3D包含大量样本文件,可以帮助您学习如何建立和研究分子模型,包括生物和无机结构。要打开示例文件,请转到“文件”>“示例文件”,然后从以下组中选择一个文件:
使用绑定工具更改绑定顺序
要使用辅助工具来更改债券定单:
1.选择目标工具(不同顺序)。
2.单击一个原子并将其拖动到另一个原子。
使用命令更改绑定顺序要使用命令更改绑定顺序:
1.右键单击进行绑定。绑定将变为黄色。
2.指向设置债券定单,然后选择债券定单。
通过更改键的最末端原子的类型来更改键顺序
此方法仅适用于将较高的键序列更改为较低的键序列。如果应用此方法将较低的键序列更改为较高的键序列,则在形成单键后,氢价完成。通过更改键两端的原子类型来更改键的顺序
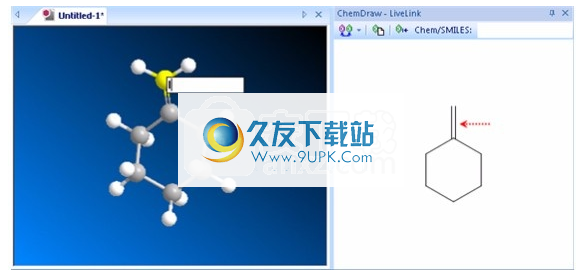
1.单击文本生成中的工具A。
2.单击连接到要更改其顺序的键上的原子。
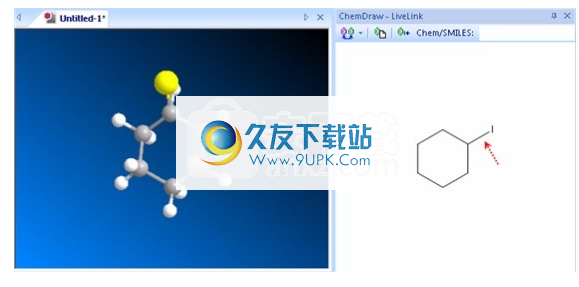
3.输入将替换所选原子的新原子类型。在以下示例中,请佩戴碘(I)而不是碳(C)。
4.按Enter。更改存储区域的键将选择新的原子类型。
通过接近键合
彼此之间指定距离内(键附近)的原子可以自动键合。
ChemBio3D根据其直角坐标和标准键长测量结果确定两个原子是否相邻。
彼此之间的距离小于标准键长加上一定百分比的原子对被认为是最接近的。百分比值越低,原子必须越接近标准键长才能被认为是最接近的键长。标准粘合长度存储在“粘合拉伸参数”表中。
设置百分比值:
1.转到文件>模型设置。出现“模型设置”对话框。
2.选择“模型构造”选项卡。
3.在以下情况下,使用“接近附加百分比的键值”滑块调整添加到标准键长度的百分比。
ChemBio3D评估原子对的邻近性。
您可以在0到100%之间调整值。例如,如果值为50,则如果两个原子之间的距离不超过连接它们的键的标准长度的50%,则认为这两个原子是最接近的。如果值为零,则只有两个原子之间的距离不大于连接它们的键的标准键长,才认为它们相邻。
在相邻原子之间创建键:
1.择自动
m在两者之间建立联系。
2.转到结构>债券。
如果两个选定原子最接近,则会创建一个键。
设定测量
建立模型时,ChemBio3D使用一组标准的长度和角度测量值来确定原子相对的位置。您可以根据需要覆盖标准度量。
要查看模型中使用的标准尺寸,请转到“结构”>“尺寸”>“全部生成”(关键长度或关键角度)。标准测量值显示在“测量”表的“最佳”(或“平衡”)列中。要在模型中编辑测量值,请在测量表的“实际”列中替换所需的值,然后按Enter。
设置密钥长度
您可以设置两个共享共价键,离子键或氢键的原子之间的长度。
设置两个原子之间键的长度:
1.选择两个原子
2.转到结构>测量>显示距离测量。出现测量表,显示原子之间的距离。
3.单击并在“实际”列中编辑值。
4.按ENTER。
设定键角
设置粘合角度:
1.选择三个连续的原子定义角度。
2.转到结构>测量>显示粘结角度测量。出现测量表,显示角度值。
3.单击并在“实际”列中编辑值。
4.按ENTER。
设定二面角
设置二面角:
1.选择定义二面角的四个连续原子。
注意:当关键点旋转时,您选择的第一个原子将移动。
2.转到结构>测量>显示二面体测量。出现测量表,显示角度值。
3.单击并在“实际”列中编辑值。
4.按ENTER。
建立密切联系
不共享键的原子被认为是紧密接触的。
设置两个紧密接触的原子之间的距离:
1.选择两个不共享键的原子。
注意:设置选定原子之间的距离时,最后一个选定原子将移动。
2.转到结构>测量>显示距离测量。出现测量表,显示两个原子之间的距离。
3.单击并在“实际”列中编辑值。
4.ENTER。
注意:您还可以使用“移动对象”工具移动原子。测量表将自动更新以反映其新位置。
原子运动
更改测量值时,您选择的最后一个原子将移动。 ChemBio3D重新定位与移动原子连接的原子,并排除与其他选定原子连接的原子。
如果测量中的所有原子都成环,则将按以下方式生成一组移动原子:
只有一个描述了所选测量原子的运动。
如果设置两个原子之间的键长或键长,则所有与固定选定原子键合的原子都不会移动。绑定到移动原子上的任何原子都会移
如果选中了“模型构造”选项卡中的“校正”复选框(转到“文件”>“模型设置”,然后选择“模型构造”选项卡),则还可以相对于移动的原子重新定位原子位置。 。
例如,考虑以下结构:

创建模型后,可以通过以下三种方式在Kekule和非本地化绑定之间切换:在模型窗口中,键入CTRL +K。转到“查看”>“模型显示”>“非本地化键”,然后选择一个选项。 。转到“文件”>“模型设置”>“模型显示”:tabandselect(或取消选择)将非本地化的键显示为虚线。设置选择颜色默认情况下,选定的原子和键以黄色突出显示。您可以在“模型设置”对话框中更改默认选择颜色。
如果键角C(1)-C(2)-C(3)设置为108度,则C(3)成为运动原子。 C(1)和C(2)保持不变。 H(11)和H(12)之所以运动,是因为它们与运动的原子键合。如果选中“自动校正”复选框,则H(10)可能是
移动,因为它是一个整流原子,并且相对于C(3)定位。
设定约束
您可以通过设置约束来覆盖ChemBio3D用于定位原子的标准测量。您可以对特定的键合长度,键合角度,二面角或非键合距离使用约束。当您使用“清理”或执行对齐,覆盖或MM2时,将应用约束而不是标准测量
计算。
要设置约束,请在“度量”表的“最佳”字段中输入一个新值。
对于二面角和非粘结距离,约束条件可以使测量保持恒定(或几乎恒定),而模型的其余部分可以通过计算进行更改。约束不会从计算中删除原子。
原子和建筑物类型
结构类型定义了模型键长,键角及其所具有原子的相对大小的结构。默认情况下,当您使用一组预定义的建筑类型构建模型时,ChemBio3D会分配建筑类型。但是,您也可以创建自己的建筑类型。
建筑类型定义模型的结构方面,原子类型定义属性,例如键能,热属性和其他非结构属性。力场使用此数据来计算模型的属性并预测模型的行为。
力场计算将考虑模型中每个原子的类型,原子的位置以及该原子所属的官能团。例如,羧基碳原子不同于烷基碳原子类型
关于正确的原子类型。正确的原子类型确定在构造时是否将原子类型分配给每个原子。原子类型(例如C Alkane)指定原子的化合价,键长,键角和几何形状。
建筑类型特征
原子的特征必须匹配这些类型特征,以便ChemBio3D将建筑物类型分配给原子。
原子符号。
绑定类型(如果为建筑物类型指定)。
绑定顺序(如果指定了绑定类型)。
双键,三键和去域键的数量。
注意:为了比较键的顺序,可以将包含一个双键的建筑类型分配给包含两个离域键的原子,例如在苯中。
如果指定了建筑类型的最大环尺寸字段,则原子必须在该尺寸的环中或更小以分配相应的建筑类型。
如果一个原子的键合数量少于建筑物类型几何体指定的配体数量,但指定了整流类型,则可以将该原子分配给建筑物类型。该报价充满了整流原子。
例如,考虑乙酸的结构类型:
O(3)符合为建筑类型O Carbonyl指定的标准。具体地,它被标记为“ O”,并且通过双键与C碳原子连接;而且,它恰好与没有三键的双键相连。
如果可以将原子分配给多种建筑类型,则按以下顺序分配建筑类型:
1.建筑物类型,其绑定类型已指定且与它们的更正类型不同。
2.建筑类型,其绑定类型已指定且与它们的更正类型相同。
3.未指定绑定类型的建筑物类型。
例如,在上述模型中,O(4)可能是几种建筑类型之一。首先,它可以是O Etheratom(优先级3),而无需指定其绑定类型。或者,它可以是O型醇,并且其键合类型与蒸馏型H型醇相同(上述优先级为2)。第三种可能性是O羧基,其键合类型是C羰基,精馏类型是H羧基(优先级1)。
由指定的结合类型具有与蒸馏类型不同的特性(上述优先级列表中的数字1),因此其优先级高于其他两种可能性,因此O羧基结构类型被分配给了氧原子。
改变建筑物的类型
您可以使用文本框更改建筑物类型和粘结特征。例如,您可以将烷烃更改为烯烃。
要更改某些原子的建筑物类型:
1.使用“从文本生成”工具单击碳原子。出现一个文本框。
2.按住SHIFT键,然后单击相邻的碳原子。选择两个原子。
3.在文本框中单击,然后键入C Alkene。
4.按ENTER。
更改建筑物类型和组合顺序以反映新模型。您可以指向原子和键以显示此新信息
分配建筑物类型
构建模型的最简单方法是允许ChemBio3D 14.0在构建时将构建类型分配给原子。
允许在施工期间分配建筑类型
n:
1.转到文件>模型设置。出现“模型设置”对话框。
2.在“模型设置”对话框中,选择“模型构造”选项卡。
3.选中正确建筑物类型的复选框。
4.单击确定。
如果选中“正确的建筑物类型”复选框,则在添加,删除或替换原子或键时将更正建筑物类型。
例:
1.建立甲烷模型,如下所示:

2.单击从文本生成工具。
3.单击氢原子。出现一个文本框,如下所示:

4.在文本框中键入C,然后按Enter。
如果选中了正确的建筑物类型复选框,将显示以下数字:

如果未选中“正确的建筑物类型”复选框,将显示以下数字:

如上例所示,当分配建筑物类型并替换了原子时,ChemBio3D尝试通过将有关原子的信息(例如其符号和键数)与原子类型中的每个记录进行比较来分配每个原子。类型。表。
另外,当您用另一种不同类型的原子替换一种类型的原子时,先前存在的原子的构造类型可能会改变。
定建筑物类型
在某些情况下,您可能想定义自己的建筑类型,是将其添加到建筑类型表中进行构建,还是添加到文件格式解释器中进行导入。
要定义自己的建筑类型:
1.转到“查看”>“参数表”>“ Chem3D建筑原子类型”。窗口中将打开Chem3D建筑原子类型表。
2.编辑建筑物类型,请单击要更改的单元格,然后键入新信息。
3.在表的每个字段中输入适当的数据。请确保该参数的名称在表的其他地方不重复。
4.关闭并保存表。现在,您可以使用新定义的建筑物类型。


 大象金服行情分析系统 1.0.1正式版
大象金服行情分析系统 1.0.1正式版 新华大宗行情软件 1.2官方版
新华大宗行情软件 1.2官方版 大智慧期货投资终端 7.81正式版
大智慧期货投资终端 7.81正式版 东方财富通level2决策版 8.6.1官方版
东方财富通level2决策版 8.6.1官方版 文华财经期货软件 6.7.11通用版
文华财经期货软件 6.7.11通用版 文华财经Smart一键通 5.5.1正式版
文华财经Smart一键通 5.5.1正式版 腾讯操盘手 1.8.0.5电脑版
腾讯操盘手 1.8.0.5电脑版 3Ds max2018广泛应用于广告、影视、工业设计、建筑设计、三维动画、多媒体制作、游戏、辅助教学以及工程可视化等领域,帮助你轻松制作出你想要的模型。有需要的用户欢迎来久友下载站下载~
3Ds max2018广泛应用于广告、影视、工业设计、建筑设计、三维动画、多媒体制作、游戏、辅助教学以及工程可视化等领域,帮助你轻松制作出你想要的模型。有需要的用户欢迎来久友下载站下载~  Bodypaint 3D可以帮助用户快速绘制出想要的3D图形,操作简单,功能强大,非常实用。有需要的用户不要错过了。
Bodypaint 3D可以帮助用户快速绘制出想要的3D图形,操作简单,功能强大,非常实用。有需要的用户不要错过了。  3D Image Maker通过设置左边图片和右边图片就可生成不同类型的三维立体图像了,支持jpg、png、bmp等格式,还可将三维图像保存为未压缩的位图图像。有需要的用户欢迎来久友下载站下载~
3D Image Maker通过设置左边图片和右边图片就可生成不同类型的三维立体图像了,支持jpg、png、bmp等格式,还可将三维图像保存为未压缩的位图图像。有需要的用户欢迎来久友下载站下载~  3d-tool可用于分析没有CAD数据的3D模型,也可以通过智能扫描模型提供对应部分的尺寸、表面积和体积,实现空间上的距离、角度和半径测量,这对于设计人员来说是不可多得的功能。除此之外,3d-tool破解版还支持SolidWorks...
3d-tool可用于分析没有CAD数据的3D模型,也可以通过智能扫描模型提供对应部分的尺寸、表面积和体积,实现空间上的距离、角度和半径测量,这对于设计人员来说是不可多得的功能。除此之外,3d-tool破解版还支持SolidWorks...  3D-boxmaker软件功能强大,简单易用,可以帮助用户更轻松快捷的设计三维包装,非常的高效实用。软件主要就是进行三维包装设计,如:电子书封面、书籍、电子杂志封面、CD封面等,支持把包装盒革面图像保存在包装盒模板文...
3D-boxmaker软件功能强大,简单易用,可以帮助用户更轻松快捷的设计三维包装,非常的高效实用。软件主要就是进行三维包装设计,如:电子书封面、书籍、电子杂志封面、CD封面等,支持把包装盒革面图像保存在包装盒模板文...  QQ2017
QQ2017 微信电脑版
微信电脑版 阿里旺旺
阿里旺旺 搜狗拼音
搜狗拼音 百度拼音
百度拼音 极品五笔
极品五笔 百度杀毒
百度杀毒 360杀毒
360杀毒 360安全卫士
360安全卫士 谷歌浏览器
谷歌浏览器 360浏览器
360浏览器 搜狗浏览器
搜狗浏览器 迅雷9
迅雷9 IDM下载器
IDM下载器 维棠flv
维棠flv 微软运行库
微软运行库 Winrar压缩
Winrar压缩 驱动精灵
驱动精灵